 United Kingdom (UK)
United Kingdom (UK) This page is intended as a resource. Click here to read about our humanization service.
The early success of mouse monoclonal antibodies led to the U.S. Food and Drug Administration (FDA) approval of the first therapeutic antibody OKT3 (muromonab) in 1986, as a treatment of kidney transplant rejection. However, most mouse antibodies were shown to have limited use as therapeutic agents because of a short serum half-life, an inability to trigger human effector functions and in particular they were recognized by the patients’ immune systems as foreign proteins resulting in a human anti-mouse antibody (HAMA) response.
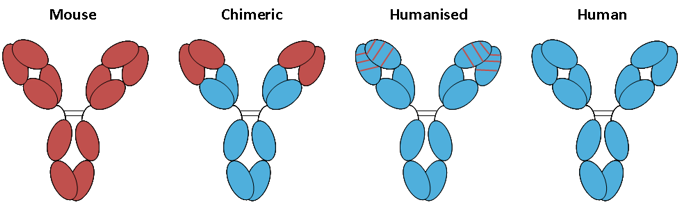
Figure. Progressive humanization of antibodies.
A schematic representation of the advancement from fully mouse antibodies, represented by red domains, to fully human antibodies, represented by blue domains.
In an attempt to reduce the immunogenicity of the mouse antibodies, genetic engineering was used to generate chimeric antibodies containing human constant domains and the mouse variable domains to retain the specificity (1, 2).
This was then taken a step further by grafting of the CDRs from a mouse antibody onto a human variable region framework, creating humanized antibodies (3). The crystal structures of rat and humanized antibodies later showed that an appropriately selected human scaffold correctly supports the orientation of the CDRs (4).
The speed of approval of this new wave of therapeutic antibodies was slower than expected due to the increase in regulatory burdens and it was not until 1996 that the FDA approved the first chimeric antibody ReoPro (abciximab), which lessons the risk of blood clots in patients with cardiovascular disease by binding to a receptor on platelets (5), and the first humanized antibody Zenapax (daclizumab) in 1997, which is an anti-CD25 antibody used to prevent organ transplant rejection (6).
The drive to reduce immunogenicity by decreasing the mouse content of monoclonal antibodies inevitably culminated in the creation of so-called fully human antibodies, which was made possible by the expression of isolated human variable domain genes in E. coli (7, 8). Two of the most widely used techniques developed for the production of fully human monoclonal antibodies are phage display, where a library of human antibodies is expressed on the surface of phage and subsequently selected and amplified in E. coli (9), and transgenic mice expressing a human antibody repertoire (10).
The image above shows a representation of mouse, chimeric, humanized and human antibodies. The chimeric, humanized and fully human monoclonal antibodies are much less immunogenic than the original mouse antibodies but human anti-antibody responses have still been noted in patients. These images are somewhat misrepresentative as immunoglobulin sequences are highly homologous across species, thus meaning a so-called fully mouse antibody is still relatively close in sequence to a fully-human one. Removal of the mouse constant domains clearly removed the most immunogenic portion of the antibody but it is argued by some that subsequent humanization has had little or no effect on the immunogenicity of therapeutic antibodies (11) and certainly only leads to marginal gains in sequence homology to a supposedly fully human antibody (12). More recent evidence suggests that while the effects of humanizing the variable domains are not as dramatic as the removal of mouse constant domains there is some reduction in the severity of the human anti-antibody response to humanized or fully human antibodies (13).
<< Antibody engineering Antibody fragments >>
References
- Boulianne, G.L., Hozumi, N., and Shulman, M.J. (1984). Production of functional chimaeric mouse/human antibody. Nature 312, 643–646.
- Morrison, S.L., Johnson, M.J., Herzenberg, L.A., and Oi, V.T. (1984). Chimeric human antibody molecules: mouse antigen-binding domains with human constant region domains. Proc. Natl. Acad. Sci. U.S.A. 81, 6851–6855.
- Jones, P.T., Dear, P.H., Foote, J., Neuberger, M.S., and Winter, G. (1986). Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 321, 522–525.
- Cheetham, G.M., Hale, G., Waldmann, H., and Bloomer, A.C. (1998). Crystal structures of a rat anti-CD52 (CAMPATH-1) therapeutic antibody Fab fragment and its humanized counterpart. J. Mol. Biol. 284, 85–99.
- Lefkovits, J., Ivanhoe, R.J., Califf, R.M., Bergelson, B.A., Anderson, K.M., Stoner, G.L., Weisman, H.F., and Topol, E.J. (1996). Effects of platelet glycoprotein IIb/IIIa receptor blockade by a chimeric monoclonal antibody (abciximab) on acute and six-month outcomes after percutaneous transluminal coronary angioplasty for acute myocardial infarction. EPIC investigators. Am. J. Cardiol. 77, 1045–1051.
- Przepiorka, D., Kernan, N.A., Ippoliti, C., Papadopoulos, E.B., Giralt, S., Khouri, I., Lu, J.G., Gajewski, J., Durett, A., Cleary, K., et al. (2000). Daclizumab, a humanized anti-interleukin-2 receptor alpha chain antibody, for treatment of acute graft-versus-host disease. Blood 95, 83–89.
- Better, M., Chang, C.P., Robinson, R.R., and Horwitz, A.H. (1988). Escherichia coli secretion of an active chimeric antibody fragment. Science 240, 1041–1043.
- Skerra, A., and Plückthun, A. (1988). Assembly of a functional immunoglobulin Fv fragment in Escherichia coli. Science 240, 1038–1041.
- McCafferty, J., Griffiths, A.D., Winter, G., and Chiswell, D.J. (1990). Phage antibodies: filamentous phage displaying antibody variable domains. Nature 348, 552–554.
- Green, L.L., Hardy, M.C., Maynard-Currie, C.E., Tsuda, H., Louie, D.M., Mendez, M.J., Abderrahim, H., Noguchi, M., Smith, D.H., Zeng, Y., et al. (1994). Antigen-specific human monoclonal antibodies from mice engineered with human Ig heavy and light chain YACs. Nat. Genet. 7, 13–21.
- Clark, M. (2000). Antibody humanization: a case of the “Emperor”s new clothes’? Immunol. Today 21, 397–402.
- Routledge, E.G., Gormon, S.D., and Clark, M. (1993). Reshaping antibodies for therapy. In Protein Engineering of Antibody Molecules for Prophylactic and Therapeutic Applications in Man, M. Clark, ed. (U.K.: Pub. Academic Titles), pp. 13–44.
- Hwang, W.Y.K., and Foote, J. (2005). Immunogenicity of engineered antibodies. Methods 36, 3–10.